


Using Mendelgen for DNA Editing (Advanced Features)
Mendelgen provides all the necessary tools for vector design. In this section you will learn how to:
- Simulate digestion
- Align sequences
- Perform codon optimization
- Translation
Simulation of Digestion
The digestion simulation feature allows you to see the predicted digestion product sizes, relative to a DNA ladder, just like on an agarose gel. To use the tool, you will need to specify the digest sites.
1) Find the digest sites of interest. They will be automatically highlighted on the Sequence Map.
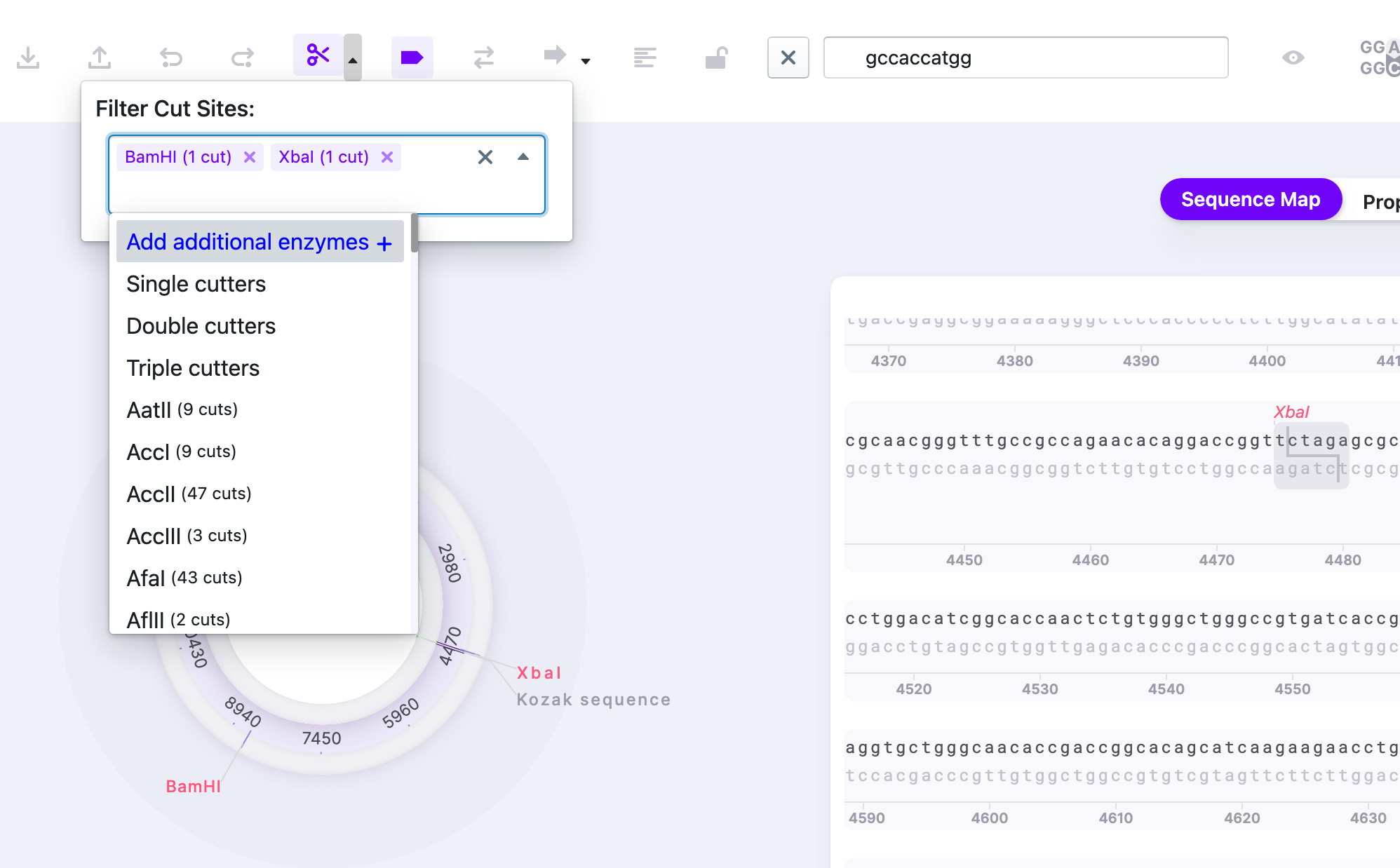
Simulate digest to see the expected products. You may choose the ladders:
- GeneRuler 1kb + DNA 75-20,000 bp
- GeneRuler 1kb + DNA 100-3000 bp
- Invitrogen 1kb + DNA 100-15,000 bp
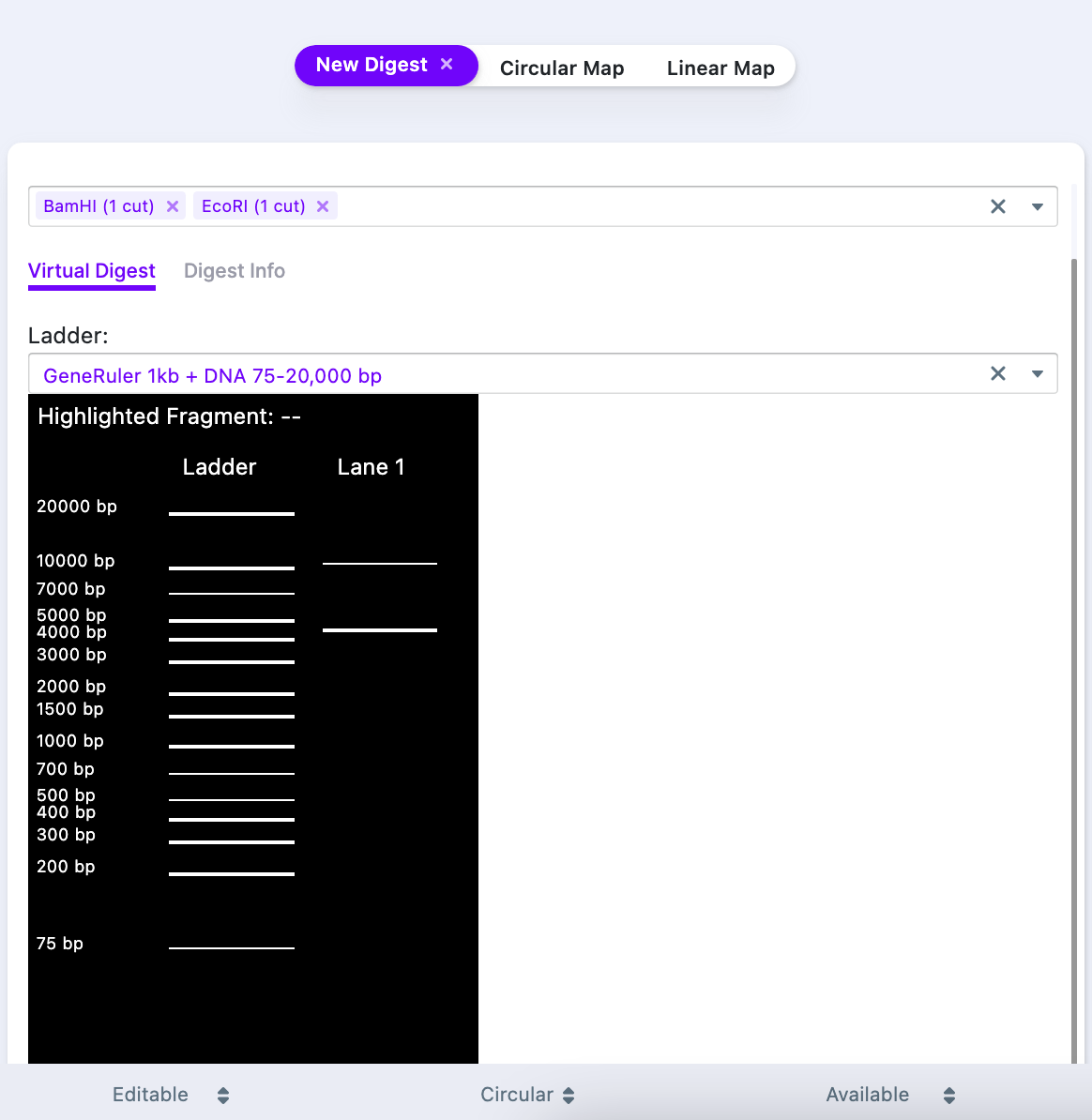
The window has two tabs. “Virtual Digest” tab shows a location of the digest fragments, alongside to a ladder of choice. The “Digest Info” contains the following data:
- Start and end of the fragment
- Length of the fragment (in base pairs)
- Left and right overhangs
- Cutter name
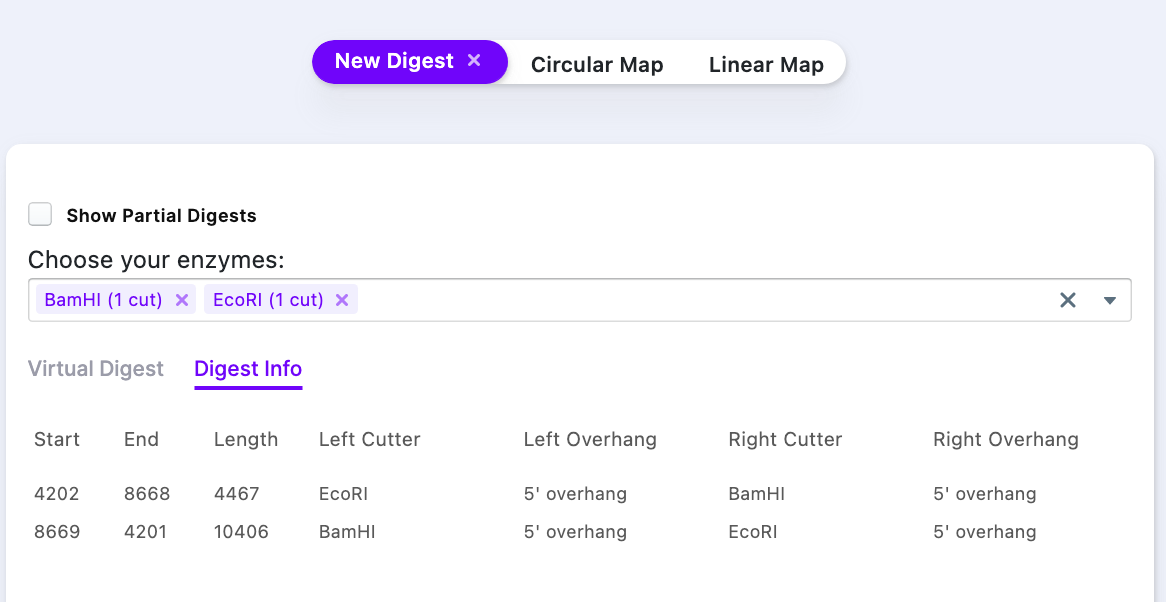
The right click on the row sends you to the location of the cut site on the Sequence Map and the cut fragment is selected automatically.
Designing Primers
To design primers (for sequencing, Gibson assembly, etc.), select a region of sequence to serve as a template for your future primer. You can see in real-time, as you select the bases, what is the GC content (in %), melting temperature, and length of the primer. Please pay attention to these values to create the best primer for your purpose. For example, the best PCR primers should correspond to the following parameters:
- Be unique (not complementary to any other region of DNA, except for the target one)
- Be long enough (usually 18-30 nucleotides). Longer primers have less binding affinity.
- Optimal melting temperature should be 65-75 C (please confirm with the annealing temperature and polymerase properties). If the melting temperature is too high, search for region with more AT content, or make the primer shorter.
- Avoid intra-primer homology, especially for longer primers.
- Target at 40-60% GC content.
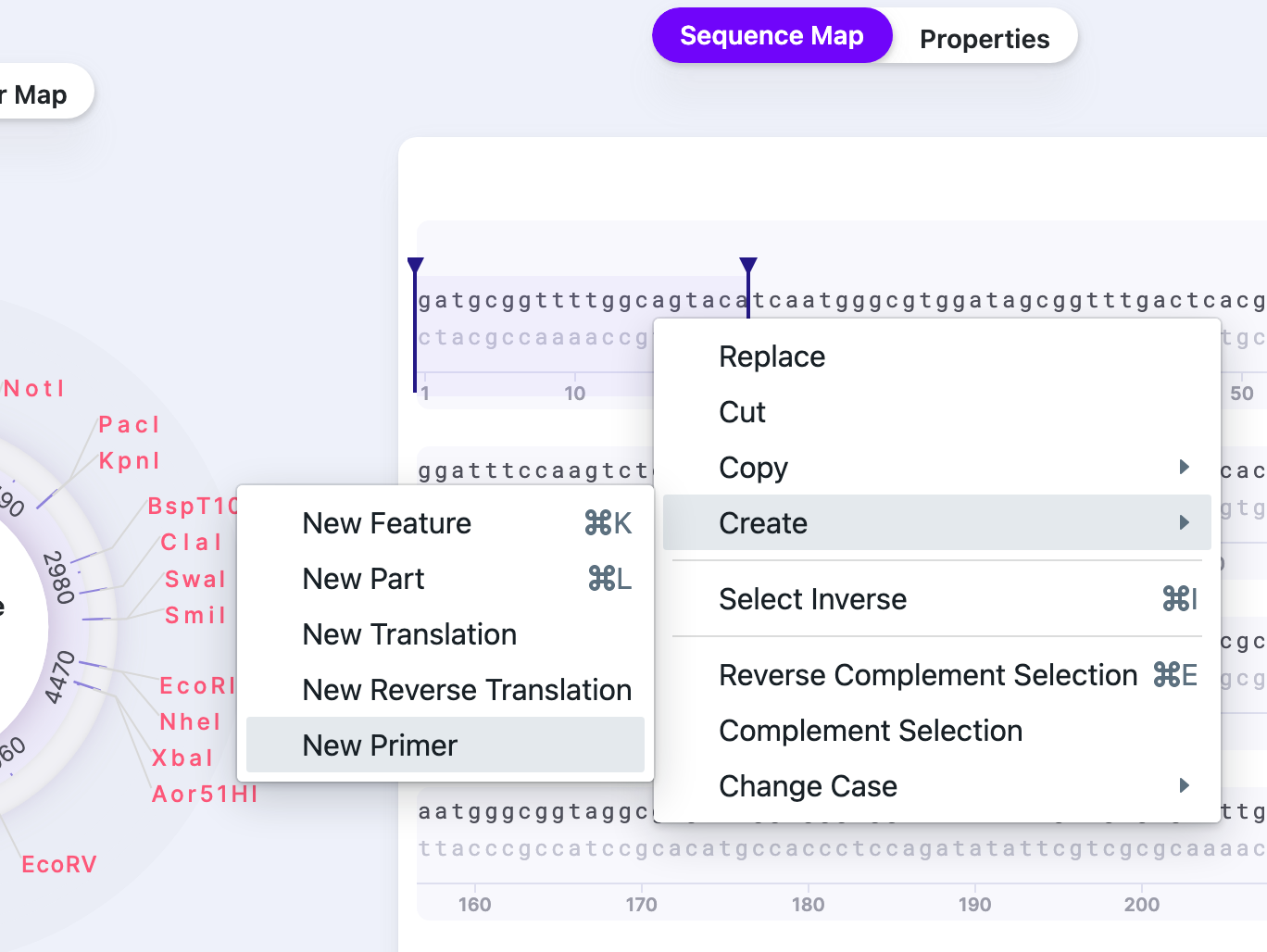
Right-click on the selection and select “Create”, and then “New primer”.
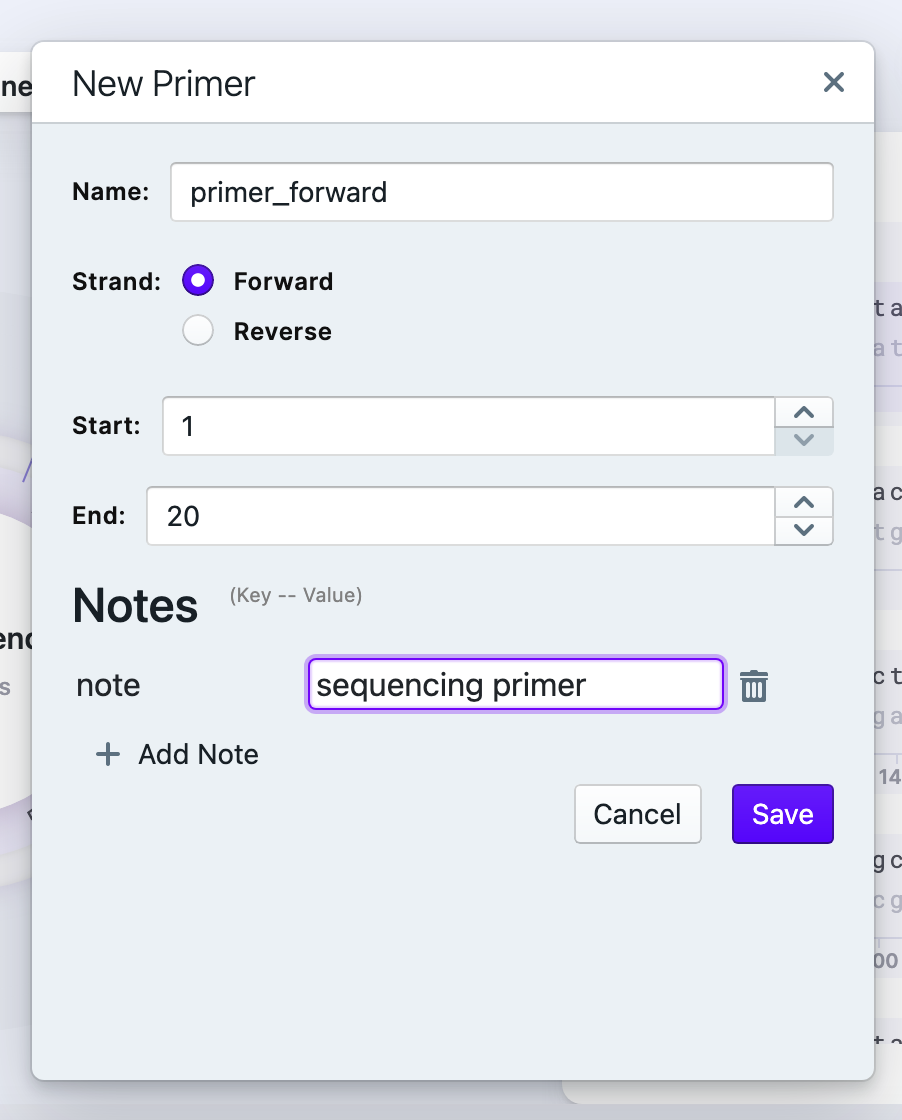
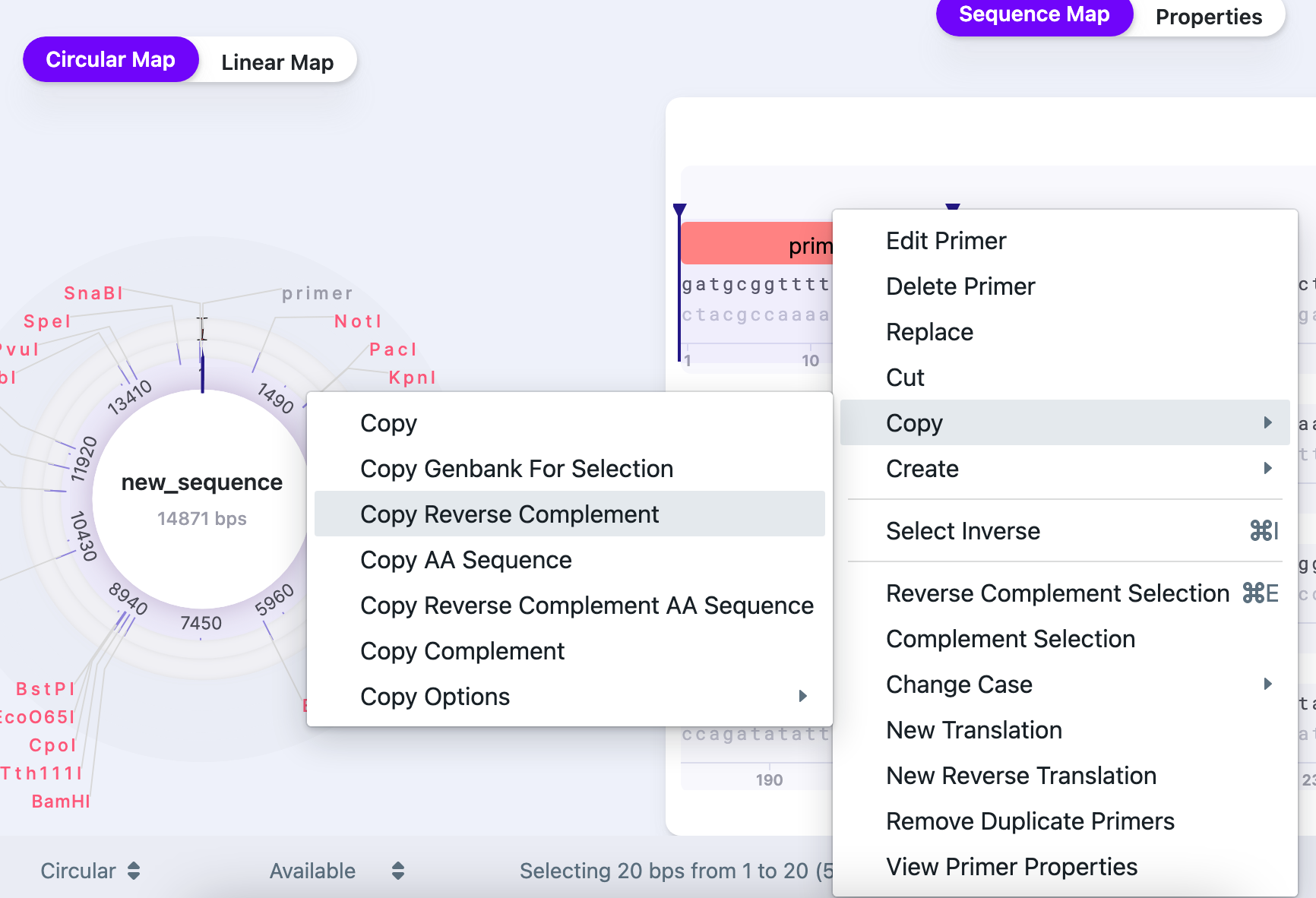
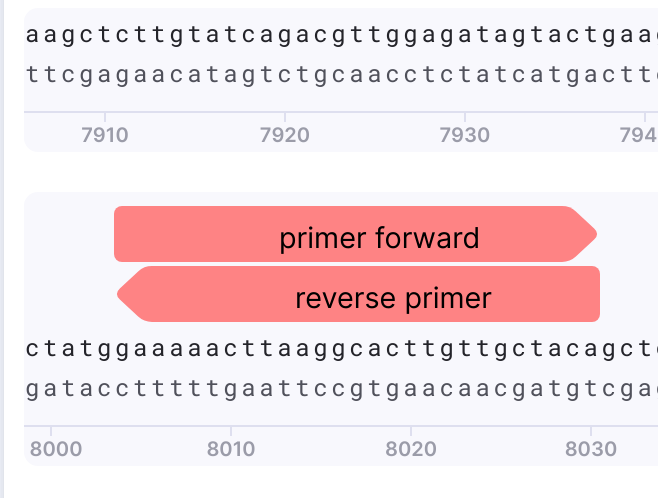
Name your primer and verify the strand. Once you created the primer the right click on it gives you options to edit, delete, replace the primer. To acquire the reverse primer, copy the reverse complement:
Sequence Alignment
For alignments, your sequence in Mendelgen will automatically become a template, however, you can change it once you add additional sequences. To get sequences for alignment, you have to upload them from your computer in .ab1, .fasta, .gb formats OR enter the sequence in plain text format.
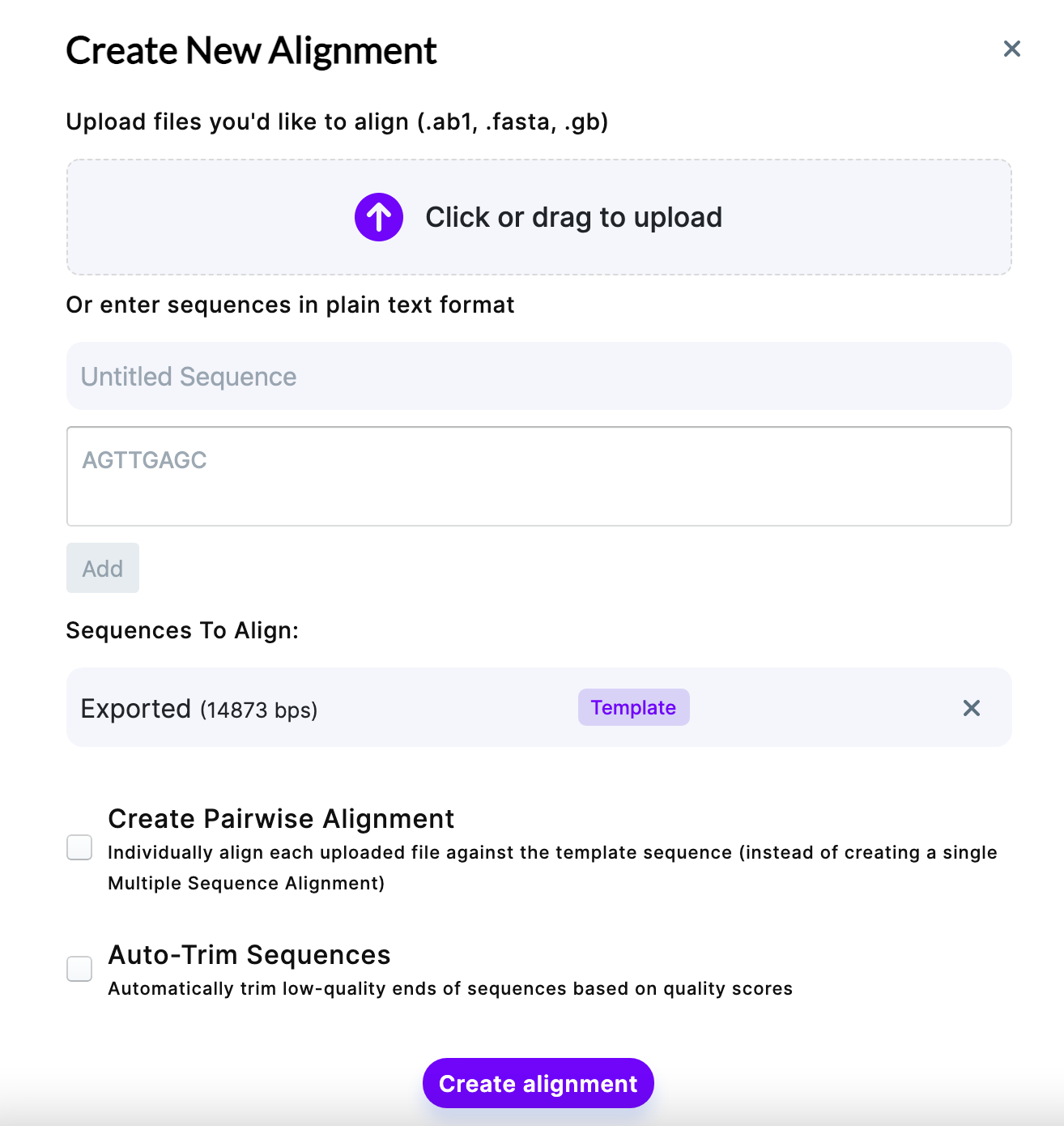
Add your sequence and name it. Once added, click on a sequence you want to make a template. You can choose to Create Pairwise Alignment (individually align each sequence against the template, if you have several files) or removing the low-quality ends of the sequence (Auto-Trim Sequences). Auto-trimming is useful, for example, when you get sequencing data back and the distant part of the sequence after primer contains very low-quality regions – Auto-Trim option allows to decrease the alignment time and simplify the further analysis by omitting those parts.
Once you verified the parameters, click “Create alignment”.
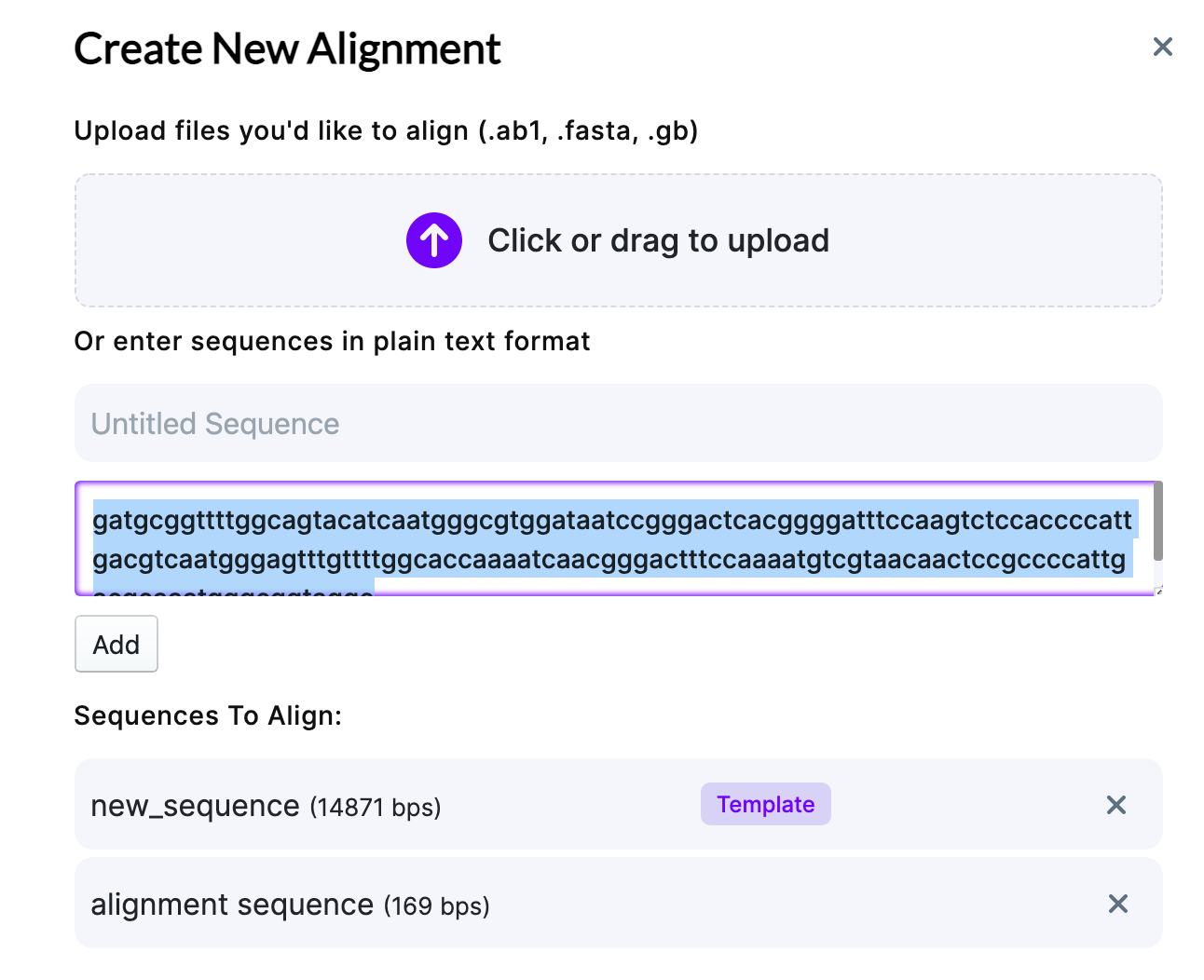
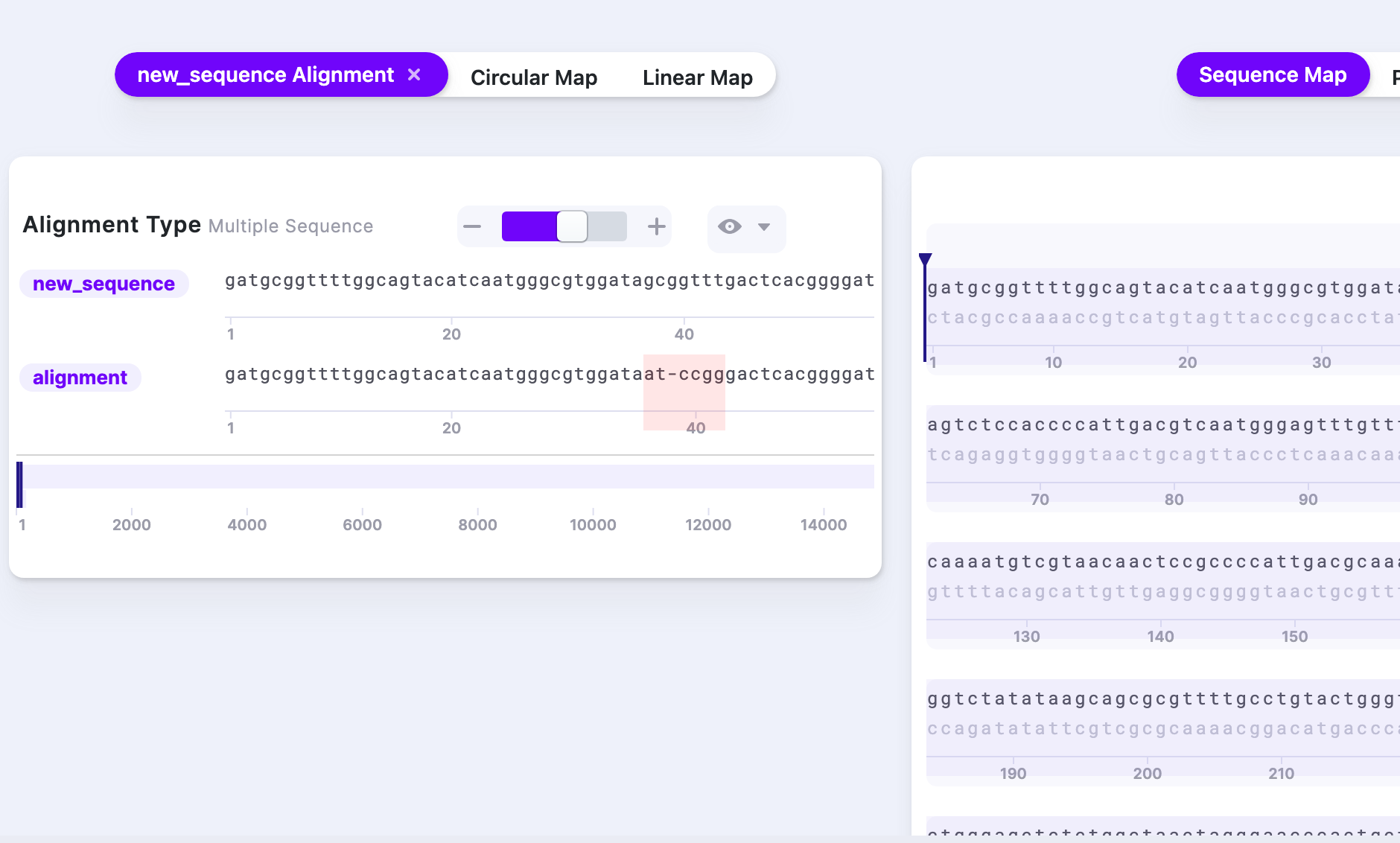
After the processing, a new tab “Alignment” appears. It shows the template and aligned sequence, with mismatches highlighted.
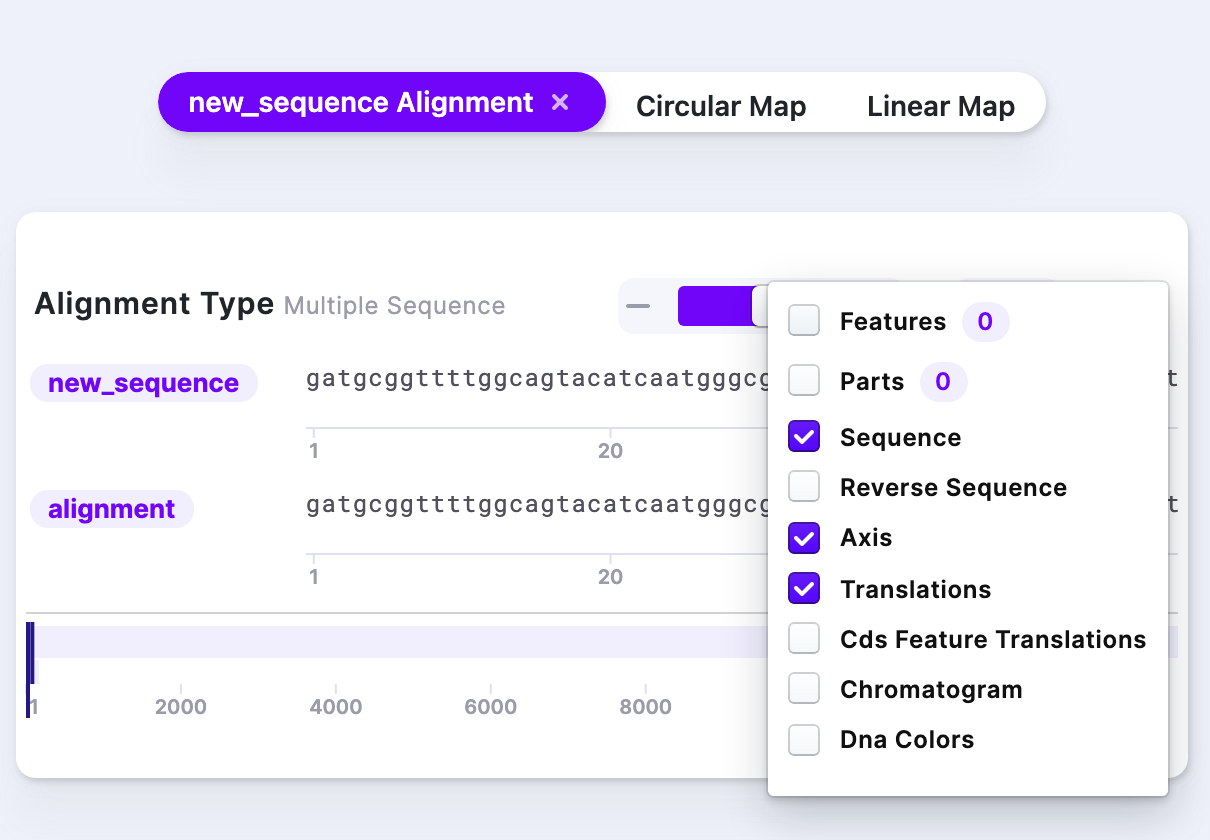
Clicking the “Visibility Options” provides additional information such as Chromatogram, DNA colors, and other display options.
Codon Optimization
The Codon Optimization tool is extremely easy to use. Use codon optimization tool to adapt your sequence to the preferred codon usage of a specific host. At the moment, Mendelgen allows you to pick from the following hosts:
- Bacillus subtilis
- Caenorhabditis elegans
- Drosophila melanogaster
- Escherichia coli
- Gallus gallus
- Homo sapiens
- Mus musculus
- Saccharomyces cerevisiae
Click on Codon Optimization from the control panel. Then, select what sequence you want to optimize on the sequence map.
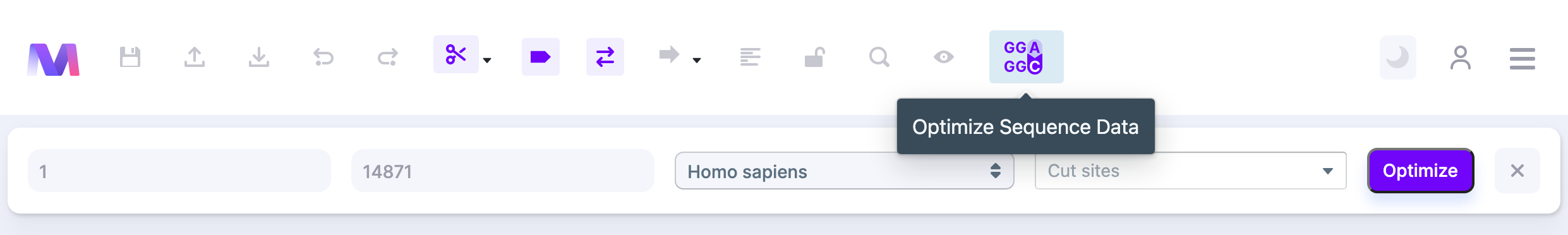
You can also pick the cut sites for optimization. You will get an error if the sequence selected is not dividable by 3.
Translation
Translation function allows to see the amino acid sequence. There are two ways to access it:
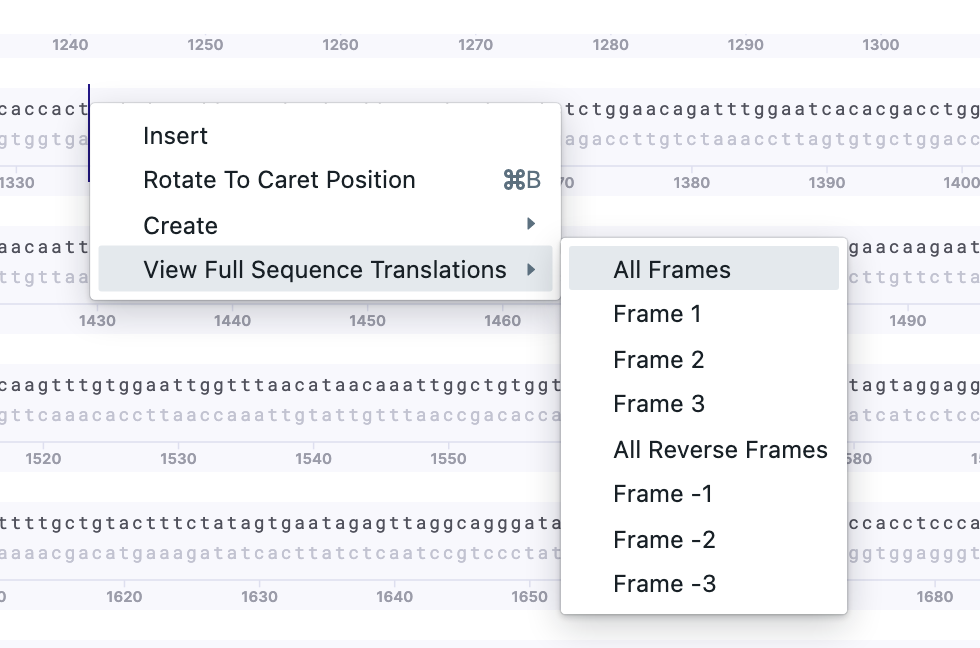
1) Full sequence translation. Right-click on the sequence – one of the options is full sequence translations. You may select frames as well.
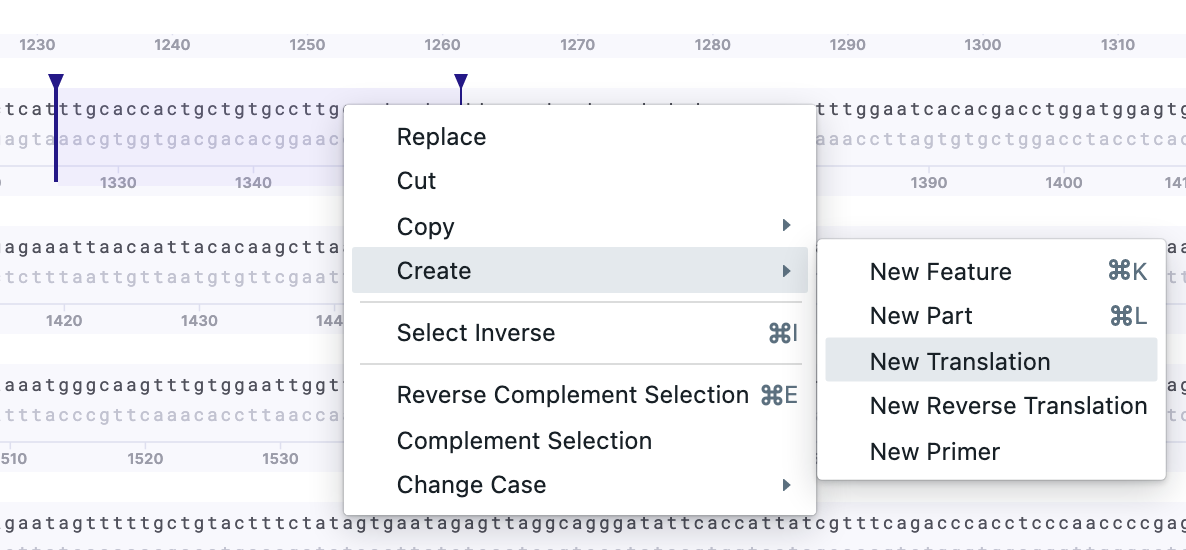
2) Partial sequence translation. Select the region of interest. Right-click – Create – New Translation/New Reverse Translation.
The additional frames will be displayed on top of each other (if selected). The sequence with all forward and reverse frames:
