


Case Study: Cloning into lentiCRISPR vector
The simplified experimental plan:
- Model the final vector.
- Digest vector. Purify backbone only.
- Design Gibson assembly primers.
- Perform PCR the fragment for insertion with Gibson primers.
- Gibson assembly reaction.
- Bacterial transformation.
- Design sequencing primers to verify insert.
- Align sequencing data to the reference sequence (final vector).
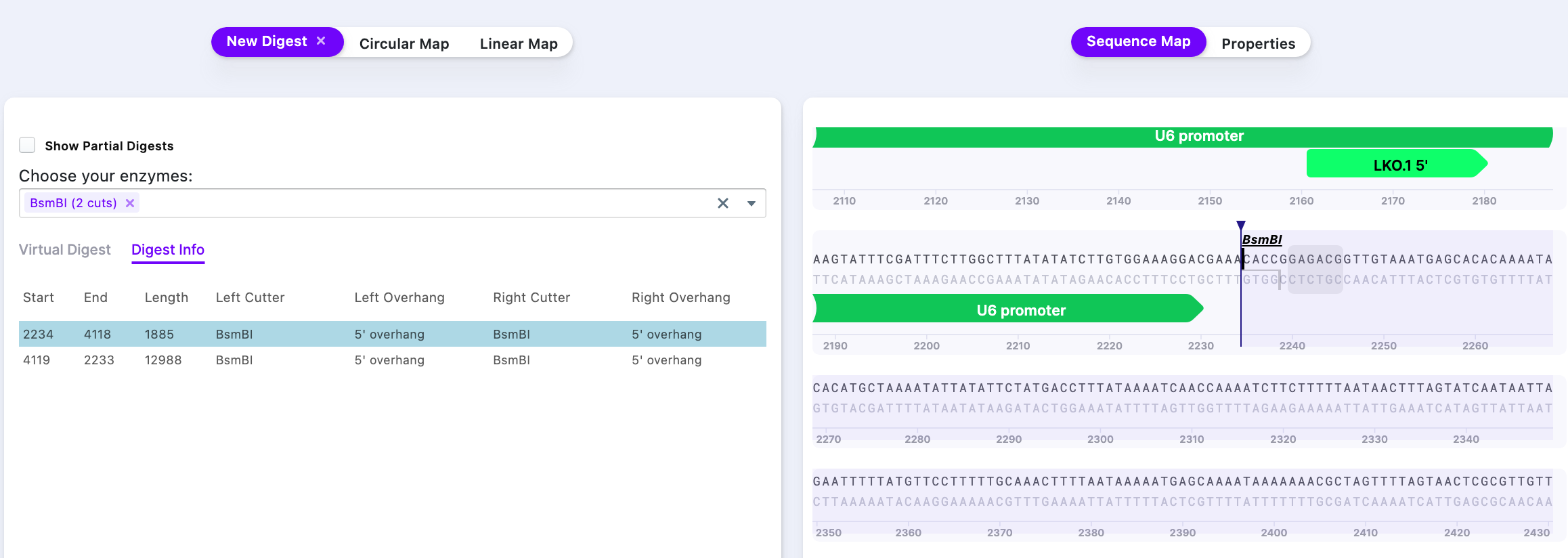
Modelling the Final Vector and Digestion. Digest vector with BsmBI enzyme. This enzyme is not available from the standard enzyme list. Add Additional Enzyme.
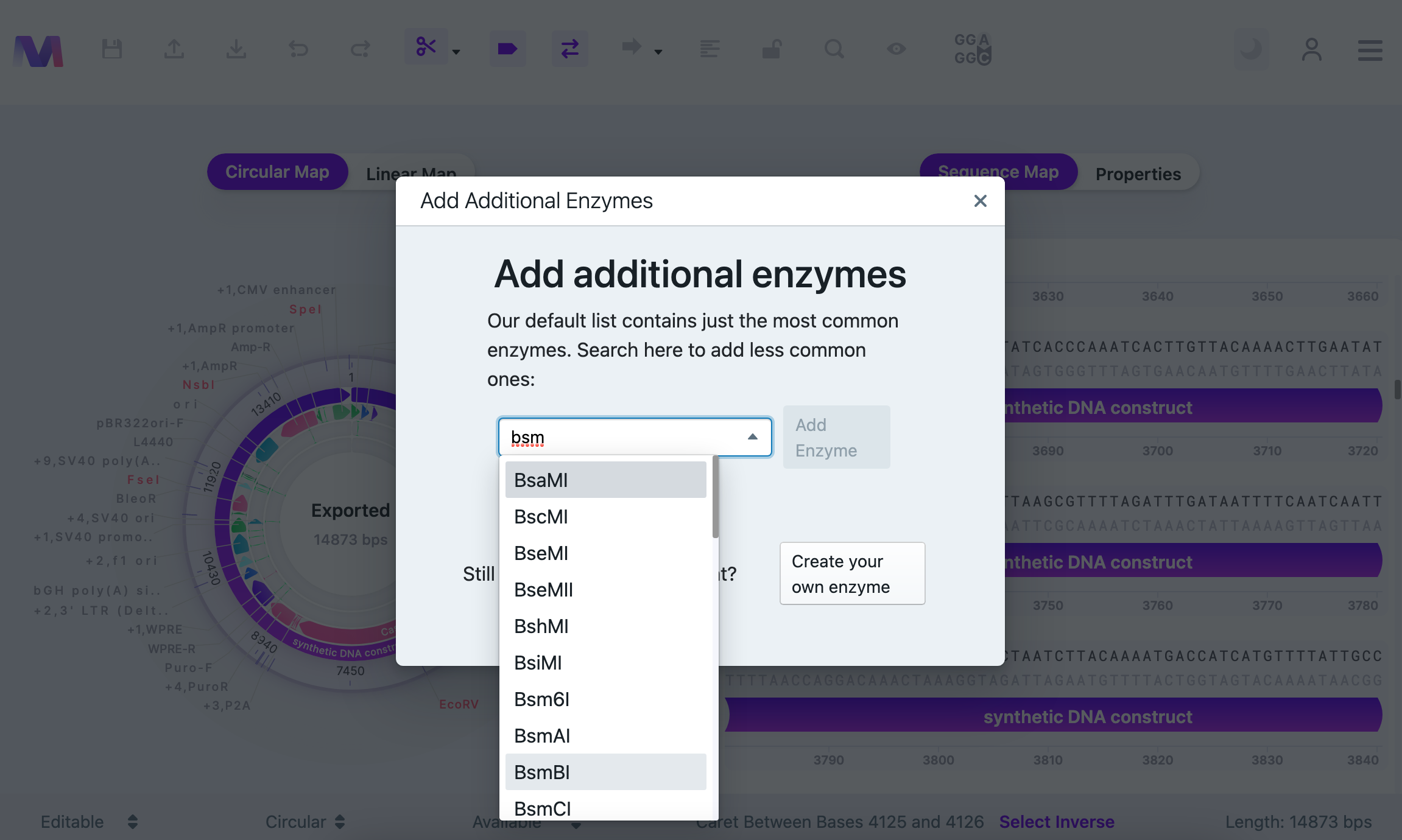
It is shown that BsmBI cuts twice in this particular vector. Run Simulate Digestion to see the digest products.
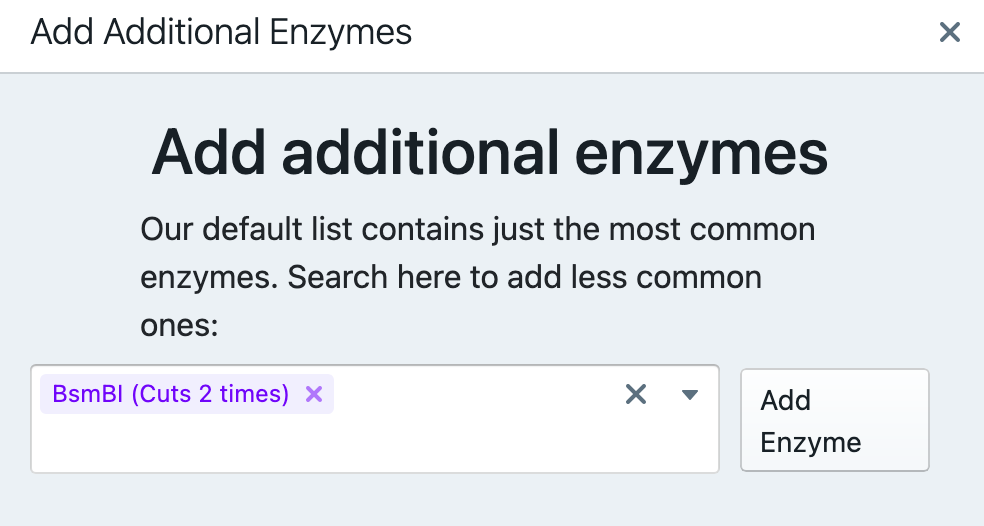
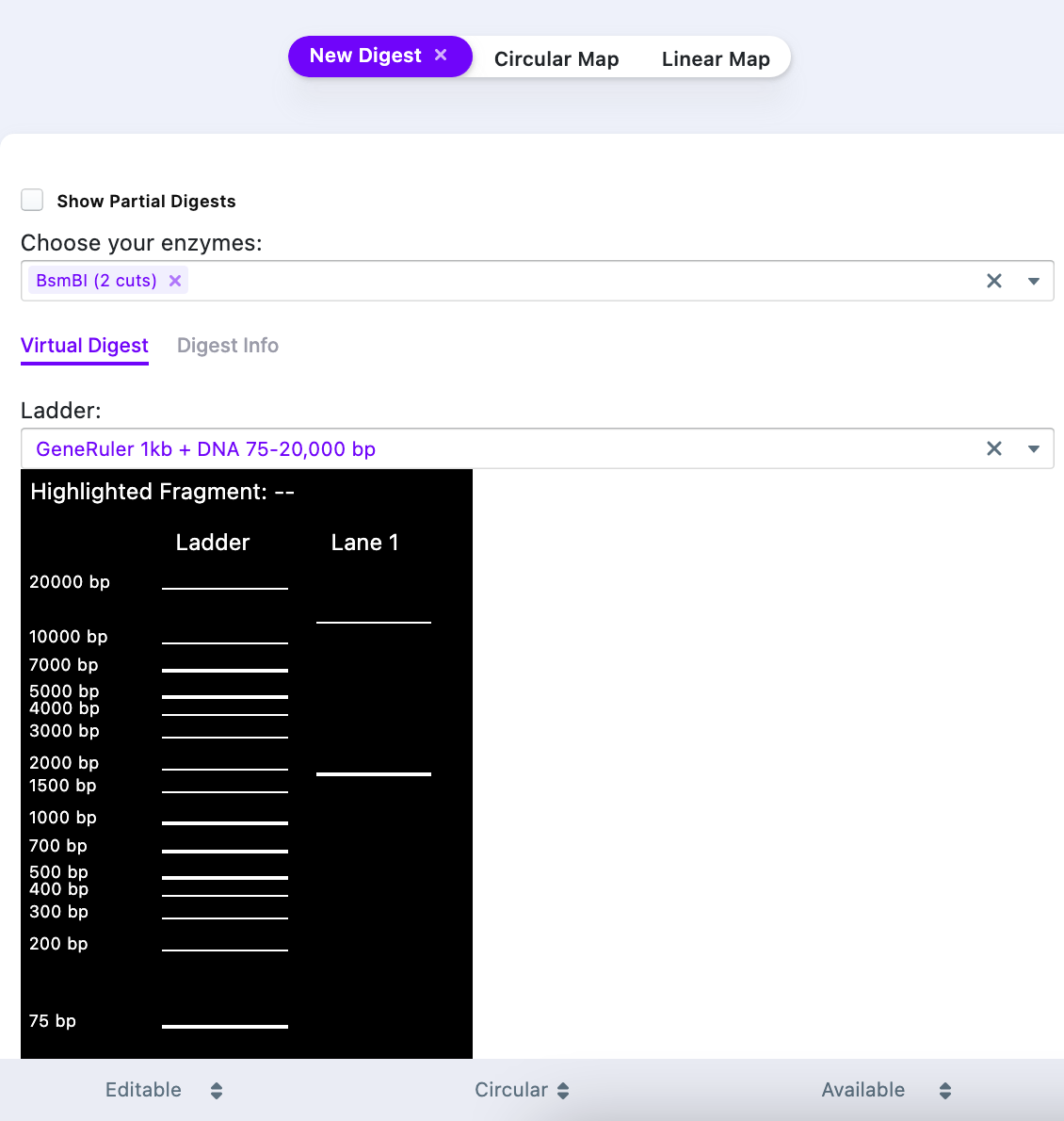
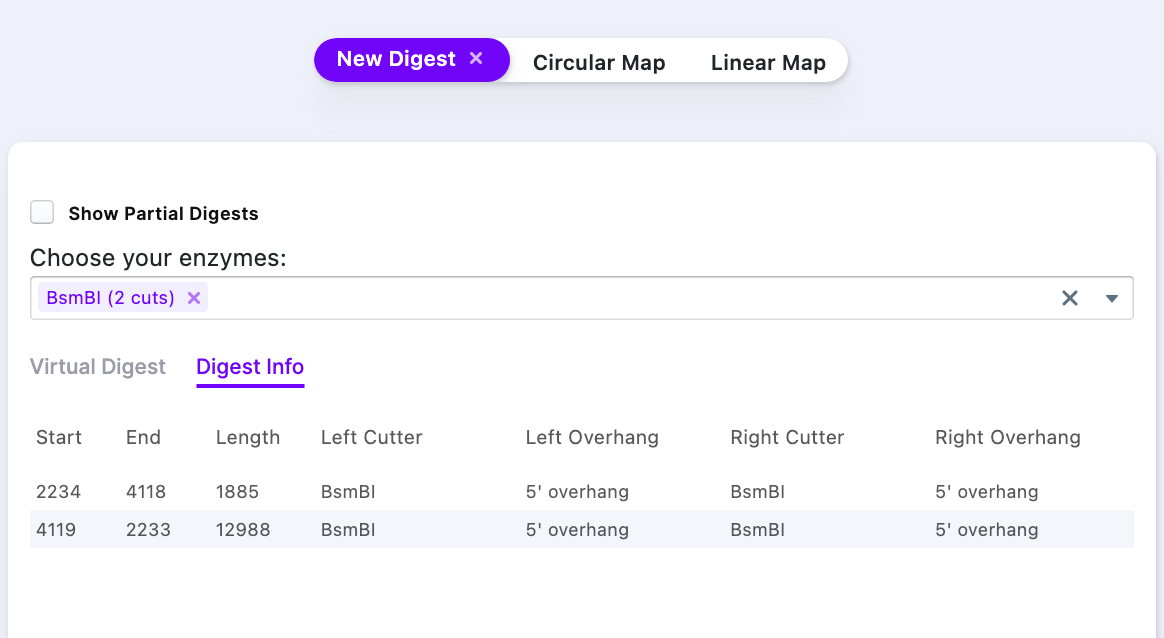
The result of the simulated digest shows that there are two products – one at 1.885 kB and another one – at 12.988 kB. To acquire vector only, it is necessary to purify (e.g., gel purification) the ~13 kB band from a gel.
The Right-click on the row with a smaller fragment size (1885 bp) selects the part of the sequence, that is being cut out. As the new sequence has to be inserted, the selected fragment has to be removed (press Delete).
Designing Gibson Assembly Primers. Designing primers for inserting the new sequence (in this case – part of the COX1 gene sequence: ctggagccggagcaccctatgtcgcagt… gagccggagcaccctatgtcaaatttccacca). In this example, the method used for vector assembly will be Gibson assembly. The following conditions were kept:
- The overhangs on the vector are 20-30 base pairs.
- The overhangs on the insert are 20-30 base pairs.
- The GC content is 40-60%.
- The Tm is 60-70C.
The general idea of Gibson Assembly is creating long primers with overhangs, complementary to both vector and insert. The overlapping fragments are digested by an exonuclease and the fragments are joined together. The required primers include forward and reverse for both sides of the insert.
First, paste the new sequence (of insert) into the vector backbone and annotate it. There are two benefits: 1) having the final vector map (after cloning). 2) using the insert as a template to design primers.
ctggagccggagcaccctatgtcgcagtatctgtctttgattcctgcctcatcctattatttatcgcacctacgttcaatattacaggcgaacatacttactaaagtgtgtaaattaattaatgcttgtaggacataataataaacgtctctgttttagagctagaaatagcaagttaaaataaggctag
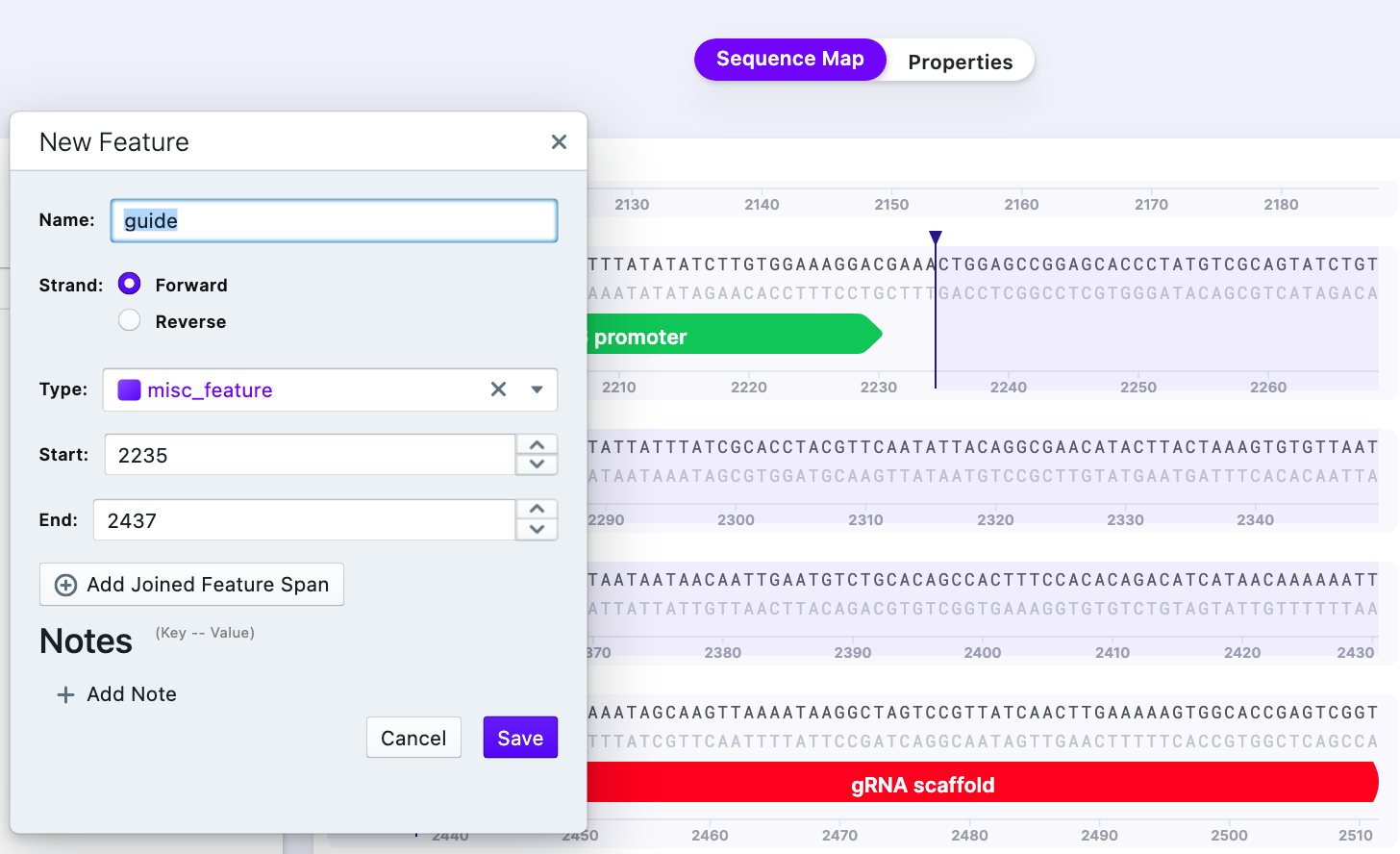
Then, select the part of the vector and insert it to serve as a primer template according to the recommendations for Gibson Assembly primer design. It is possible to annotate parts for convenience using “Create”–>”New Part”.
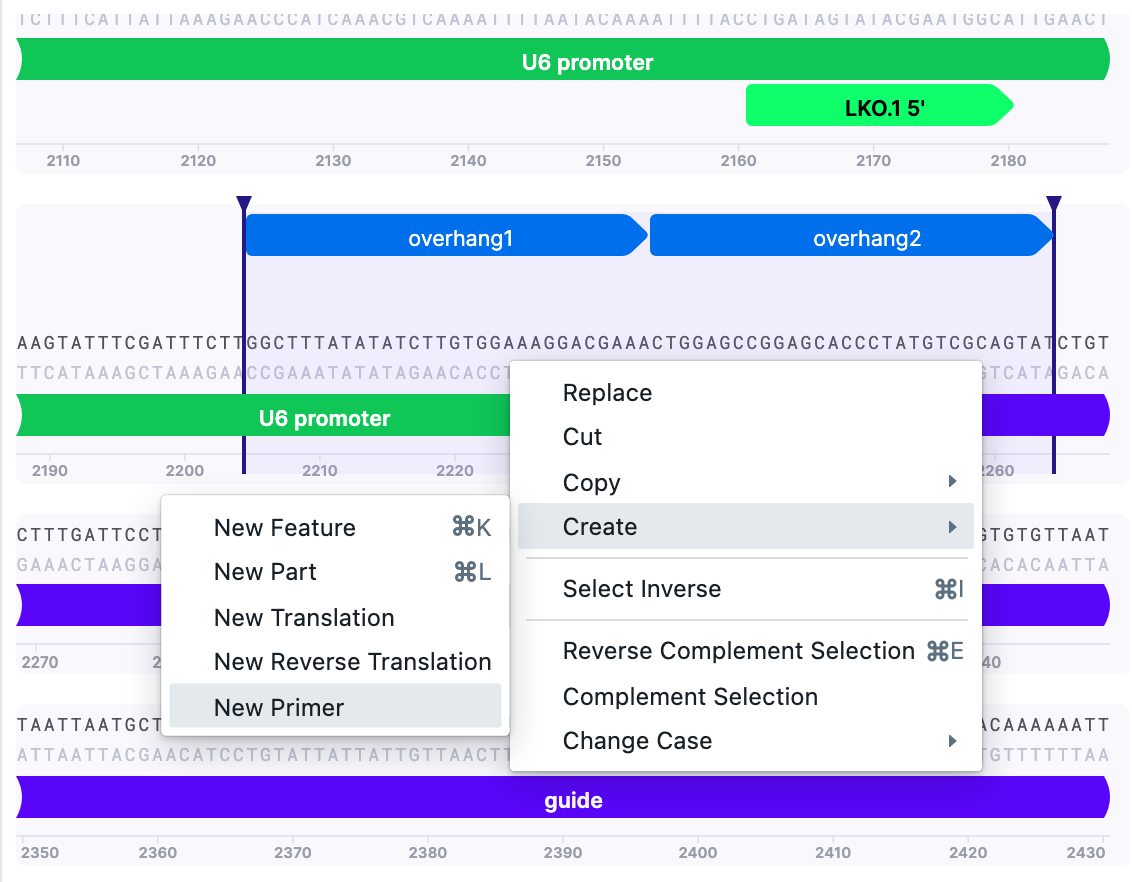
On the selected overhangs, create a New Primer:
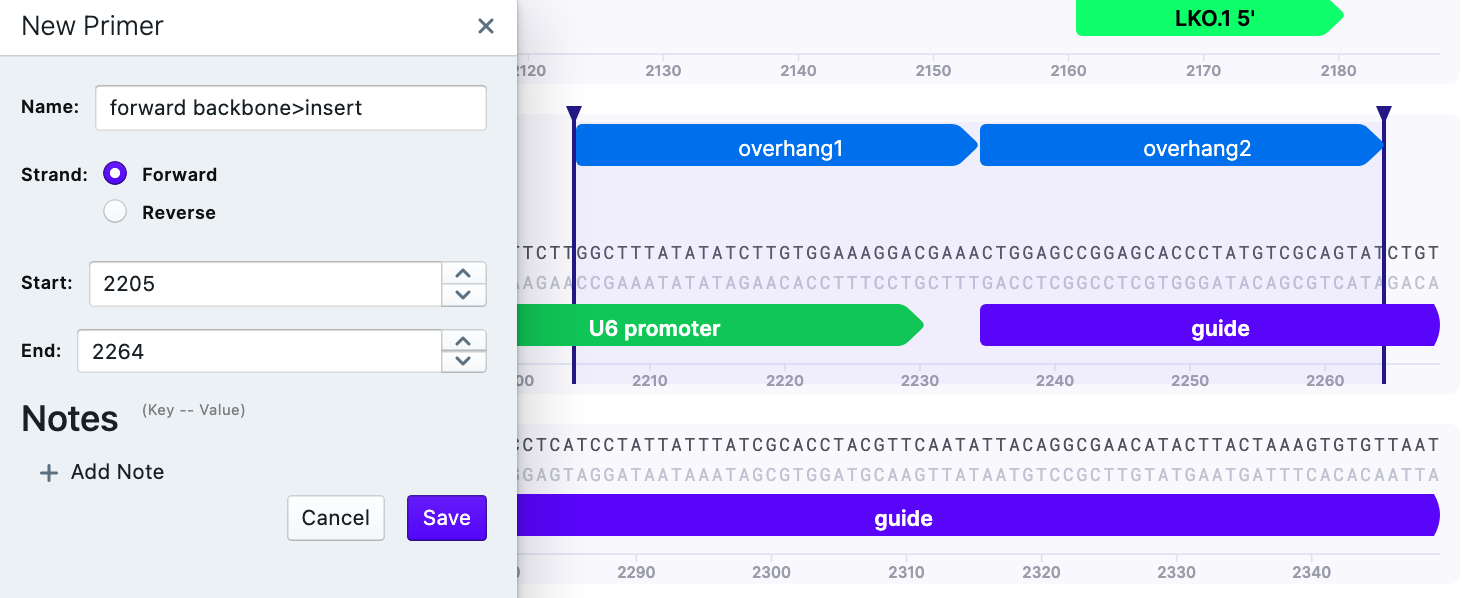
Primer 5’–3’ (backbone>insert), site A would be GGCTTTATATATCTTGTGGAAAGGACGAAACTGGAGCCGGAGCACCCTATGTCGCAGTAT
As for Gibson Assembly, one needs primers in forward and reverse direction. To save the reverse primer: Primer 3’–5’ (insert>backbone), site A would be: CTTGCTATTTCTAGCTCTAAAACAGAGACGTTTATTATTATGTCCTACAAGC
Repeat for site B.
Transformation and Sequencing Primers Design. After the primers are synthesized, PCR is done and Gibson reaction is assembled, the last step is to transform bacteria with a new vector. To verify the insert, design the sequencing primers. Each sequencing company has their own submission rules. For this case study, the primers must correspond to the following rules:
- 18-25 bp in length
- Designed with an extra “space” before the region of interest.
- GC% is 40-60%
- Tm is 60-70 C
i Designing a primer within human U6 promoter gives us a universal primer, which can be used in other experiments as well – some sequencing companies have U6 promoter primers available in the company already by default, so check its availability before ordering the custom primer.
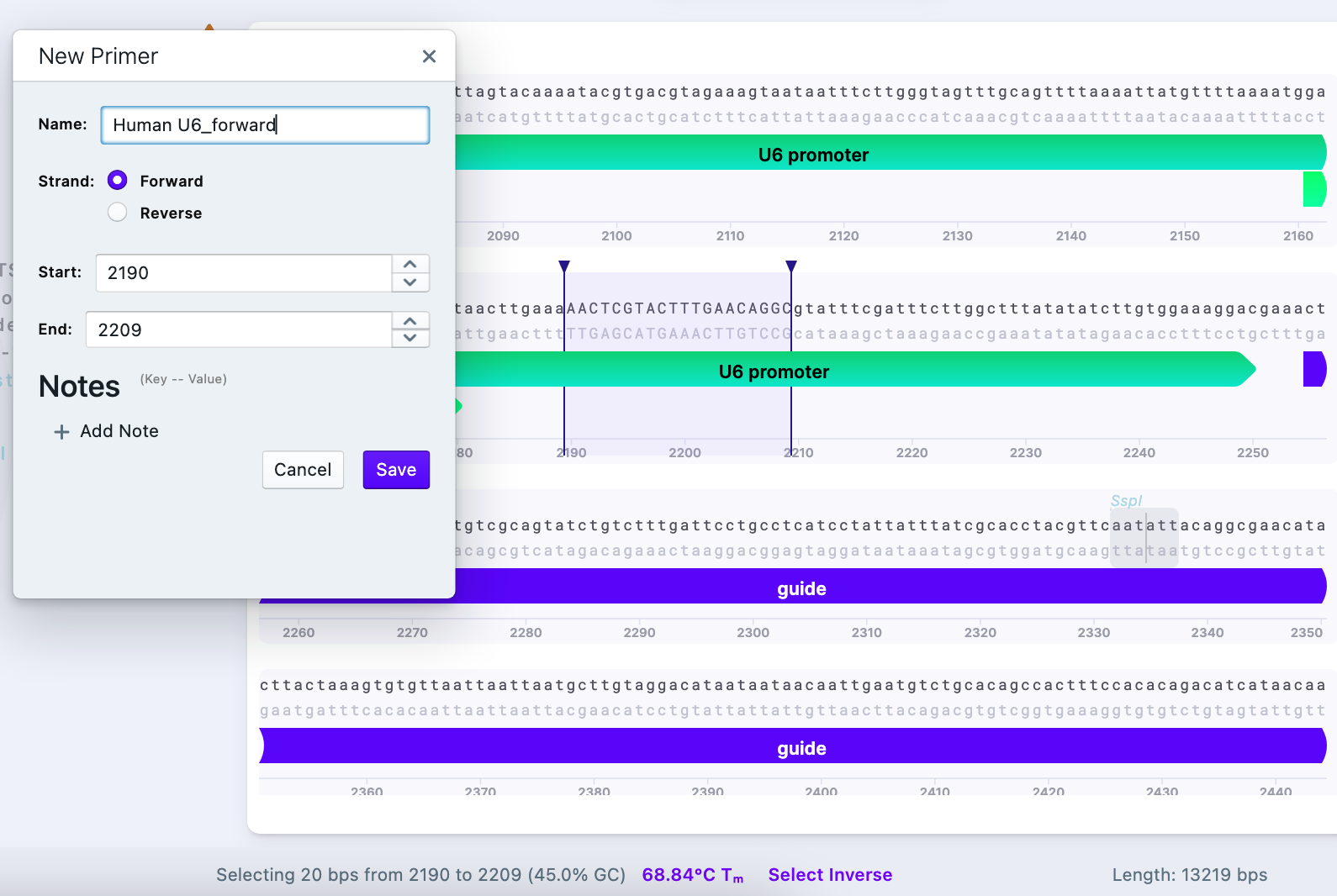
The designed primer has the following parameters:
- Length: 20 bp
- GC% = 45%
- Tm = 68.84 C
Sequence Alignment. After the Sanger sequencing results are back, align the received sequencing data to the template (a vector designed).
Go to Align to this Sequence and select the sequencing file.
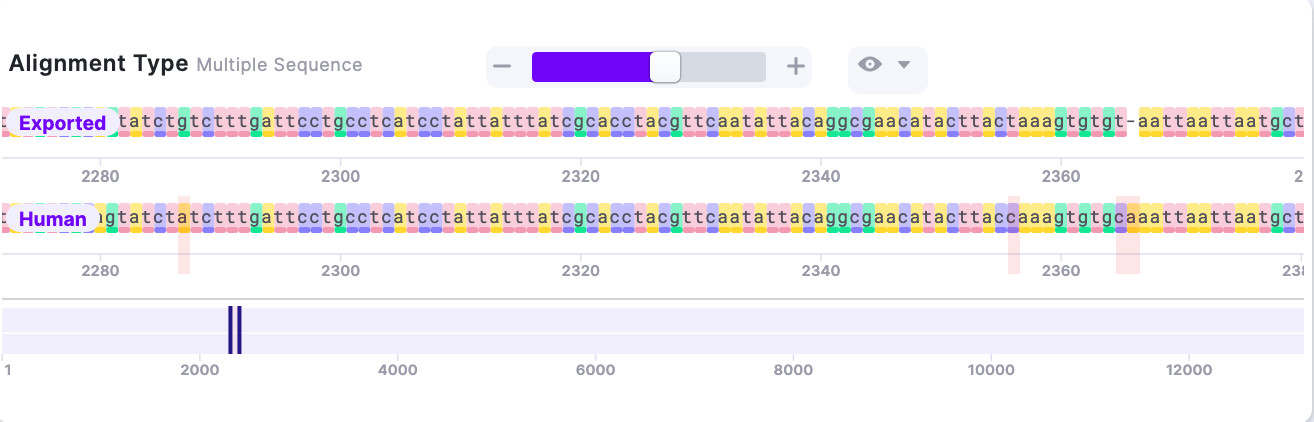
The alignment showed several mismatches.
To see if they are affecting the gene function, translate the insert’s (sgRNA’s) sequence.
Copy the AA sequence:
Template: LEPEHPMSQYLSLIPASSYYLSHLRSILQANILTKVCKLINACRT***
Sequencing file: LEPEHPMSQYLSLIPASSYYLSHLRSILQANILTKVCKLINACRT***
The AA sequences are identical; therefore, the function is likely to be preserved even with some mismatches.